
Introduzione
In quasi duemila e cinquecento anni di medicina l’umanità è riuscita a cancellare appena due malattie. La prima, nel 1980, è stata il vaiolo, mortale in un terzo dei casi. La seconda, nel 2011, e forse meno nota, è stata la peste bovina o Rinderpest – che a dirla tutta di peste aveva ben poco, essendo causata da un parente del morbillo, da cui forse il morbillo stesso è evoluto. La mortalità della peste bovina poteva raggiungere fino al 100%, e non sorprende che sia stata oggetto di preoccupazioni fin da tempi antichissimi. La quinta delle piaghe d’Egitto descritte nell’Esodo avrebbe potuto essere proprio la Rinderpest: “La mano del Signore sarà sul tuo bestiame che è nei campi, sui cavalli, sugli asini, sui cammelli, sui buoi e sulle pecore; ci sarà una tremenda mortalità.” (Esodo, 9:3) – non era infrequente che la peste bovina passasse anche ad altri ungulati a dita pari, tra cui appunto cammelli e pecore, anche se cavalli ed asini non sembravano essere colpiti. Sarà stata una svista.
Il 9 novembre del 1711, trecento anni esatti prima dell’eradicazione, un medico emiliano forniva a Venezia un resoconto sulla panzoozia (l’equivalente animale di una pandemia) di Rinderpest che aveva colpito l’Europa dal 1709 al 1720, fornendo forse la prima elegante menzione di quello che sarebbe poi diventato un proverbio italiano tanto noto quanto dimenticato. Nella sua tredicesima orazione, Bernardo Ramazzini così diceva: “Atque hæc quidem ad Therapœjam, modo pauca quædam documenta pro hujus meæ prælectionis coronide, in prophilaxeos gratiam lubet proponere, quando longe præstantius est præservare, quam curare, sicuti satius est tempestatem prævidere, ac illam effugere, quam ab ipsa evadere”. Una grossolana traduzione sarebbe: “E senza dubbio per la terapia, con solo alcuni insegnamenti a coronamento della mia lezione, vorrei mostrare la grazia nei metodi profilattici, poiché è di gran lunga più efficace prevenire che curare, così come è più saggio prevedere una tempesta e scamparla, piuttosto che evadere da essa” [1].
Trovare una cura alle malattie neurodegenerative è stato a lungo considerato uno degli obiettivi della neurologia, fin dalla descrizione della più comune forma di demenza da parte di Alois Alzheimer (1864-1915) e Gaetano Perusini (1879-1915) all’inizio del secolo scorso. Nei lavori dell’ingiustamente dimenticato Perusini troviamo due fondamentali intuizioni. La prima è l’associazione tra le cosiddette “placche senili” (accumuli di proteina beta-amiloide) e la morte dei neuroni: “La formazione delle placche senili è un evento associato all’involuzione del sistema nervoso centrale umano”. La seconda è il riconoscimento del possibile contributo vascolare alla malattia: “Rimane una questione aperta se agenti nocivi in grado di determinare la malattia agiscono solo sui vasi sanguigni o danneggiano egualmente neuroni e vasi” [2]. Ci si potrebbe chiedere il senso di questa digressione storico-campanilistica, ma la risposta è molto semplice: cent’anni dopo, le due caratteristiche chiave identificate da Perusini – gli accumuli di proteina beta-amiloide e il carico vascolare – continuano ad essere fondamentali. Ancora più interessante – e provocatorio – è il fatto che la prima paziente descritta da Alzheimer potesse non soffrire clinicamente di Alzheimer, ma di demenza vascolare. Torneremo più avanti su questo aspetto [3].
La prima presentazione della malattia che fece Alzheimer nel 1906 ad un congresso di psichiatria a Tübingen fu accolta dall’indifferenza generale, e commentata negli atti del congresso dalle parole “zu kurzen Referat nicht geeignet” – non adatto ad una breve relazione. Ad oggi, esistono interi congressi dedicati alla malattia di Alzheimer. Ricercando la parola “Alzheimer” su PubMed, il motore di ricerca più usato per gli articoli di ambito medico, si ottengono 214054 risultati in 110 anni; restringendo la ricerca solo ai primi cinque mesi del 2023, sono 7799. Se ipotizzassimo un tempo medio di due secondi per leggere solo il titolo di ciascuno di questi ultimi articoli, impiegheremmo quattro ore e mezza – nel frattempo, verrebbero pubblicati altri 10 articoli. Si stima che 150 milioni di persone al mondo soffriranno di demenza nel 2050, e la maggior parte di questi soggetti avranno la malattia di Alzheimer [4]. In effetti, l’argomento non era proprio adatto ad una breve relazione!

Come (non) funziona la malattia di Alzheimer
In quella che è la spiegazione della malattia di Alzheimer attualmente più accettata, l’aspetto centrale è l’accumulo di una (in realtà, come vedremo, due) proteina “mal ripiegata” (misfolded), avente la capacità di diffondersi tra un neurone e l’altro causandone la morte.
La storia inizia con la proteina APP, una proteina a cavallo della membrana delle cellule, che viene clivata (cioè “tagliata”) da due enzimi, chiamati rispettivamente α-secretasi e β-secretasi, prima di essere ulteriormente clivata da un terzo enzima, γ-secretasi (di cui fanno parte, tra le altre, due proteine chiamate presenilina 1 e presenilina 2). La prima di queste vie porta ad un frammento “neuroprotettivo”, la seconda ad un frammento neurotossico chiamato Aβ (o beta-amiloide) [5]. In condizioni normali, l’α-secretasi fa gran parte del lavoro, e solo il 10% dell’APP viene processata nella via amiloidogenica [6]. Per una serie di meccanismi a cascata successivi, l’Aβ è poi il trigger della modifica in senso neurotossico di un’altra proteina, chiamata tau [7].
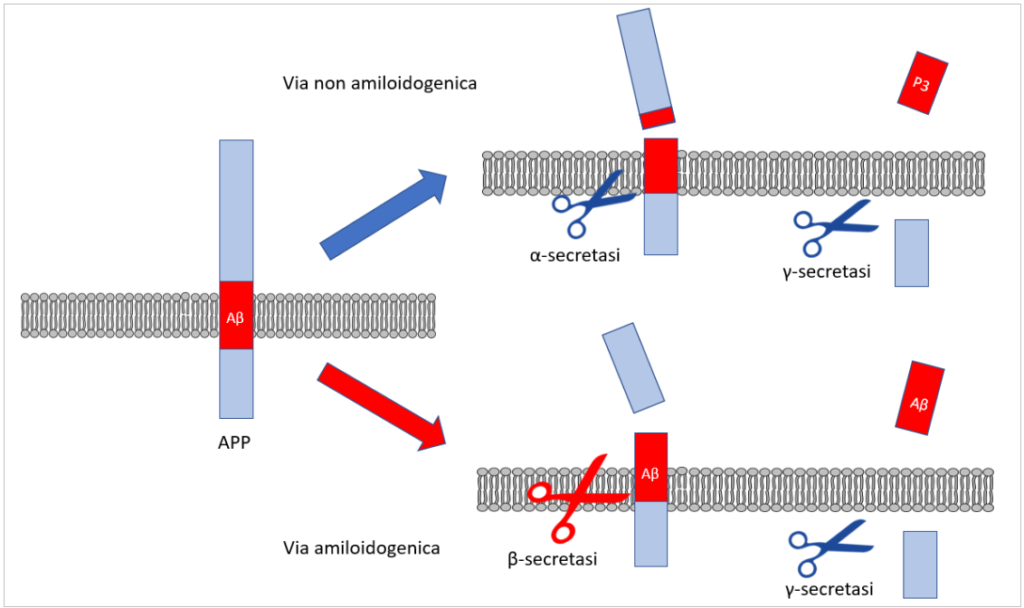
Le proteine (tra cui APP e i suoi prodotti) vengono sintetizzate dalle cellule come una semplice catena lineare di singoli aminoacidi. Nella loro trasformazione successiva possono modificarsi aggiungendo nuovi gruppi chimici (come gruppi fosfato – nel processo detto fosforilazione), e vengono ripiegate in strutture via via più complesse, da cui dipende in parte la loro funzione. Tuttavia, questo processo non è sempre effettuato in modo corretto, ed alcune proteine vengono “mal ripiegate” (misfolded proteins). Attualmente, si pensa che questo meccanismo sia alla base della maggior parte delle malattie neurodegenerative.
Per spiegare come dalla beta-amiloide mal ripiegata si arrivi alla demenza bisogna chiamare in causa ancora una volta i bovini. Negli anni ’80-’90 si affacciava sull’Europa l’encefalopatia spongiforme bovina, una malattia neurodegenerativa delle mucche chiamata popolarmente “morbo della mucca pazza”. Questa malattia è stata la causa di circa ventimila morti tra gli umani, legati esclusivamente al consumo di carne bovina “infetta”. Eppure, l’agente infettivo in questo caso non era un organismo, bensì una proteina (la proteina prionica) già nota ai neurologi per essere responsabile della malattia di Creutzfeldt-Jakob, dell’insonnia familiare fatale e del kuru – una malattia che colpiva donne e bambini della tribù Fore della Papua Nuova Guinea, derivante dal consumo ritualistico del cervello dei defunti (l’ultimo paziente è morto verosimilmente nel 2009). La proteina prionica (PrP) è un esempio di come una proteina mal ripiegata possa non solo perdere la propria funzione, ma anche acquisire proprietà del tutto nuove, come la capacità di trasmettere la propria conformazione sbagliataanche ad altre proteine della stessa famiglia (un po’ come se sbagliare a piegare una maglietta trasformasse progressivamente l’armadio in una distopia post-atomica!). Questo genera una serie di eventi a cascata, tra cui infiammazione cerebrale, morte neuronale ed atrofia, che si riflettono in quadri clinici variabili a seconda delle regioni cerebrali colpite. Inevitabilmente, il processo finisce per interessare tutto l’encefalo, portando invariabilmente a morte il paziente.
Al giorno d’oggi abbiamo due modi per valutare in vivo la presenza della patologia di Alzheimer. Uno è l’analisi dell’Aβ e della tau – nelle sue forme totale e fosforilata – nel liquido cefalorachidiano (un fluido che bagna il cervello e il midollo spinale), ottenibile tramite una puntura lombare – che al giorno d’oggi si dovrebbe effettuare con aghi atraumatici di basso calibro, riducendo sensibilmente il discomfort della procedura [8,9]. L’altro è l’utilizzo di tecniche di imaging nucleare che sfruttano composti radioattivi con affinità per l’Aβ (nel caso della PET-amiloide con PiB, Florbetaben, Florbetapir e Flutemetamol) o tau (nel caso della PET-tau con Flortaucipir, attualmente ancora molto poco diffusa).
Attualmente si ipotizza che l’Aβ, la tau, ed altre proteine implicate nelle malattie neurodegenerative (come l’alfa-sinucleina nel Parkinson) si comportino in un modo molto simile alla proteina prionica, aggregandosi in strutture via via più complesse e propagandosi difficili da rimuovere, che alla fine portano alla morte neuronale. Si parte dai monomeri (ovvero le semplici proteine di Aβ), per arrivare via via ad oligomeri, protofibrille, fibrille e infine alle placche – quelle già descritte da Perusini. Tra queste, oligomeri e protofibrille sono solubili, mentre le fibrille sono insolubili, e si depositano nei neuroni, diventando molto difficili da rimuovere. Vedremo in seguito come tutto ciò sia rilevante per le nuove terapie. Questo processo parte da certe aree relativamente specifiche per ciascuna proteina, il che determina il fenotipo clinico della malattia – per esempio, un accumulo in sede ippocampale nella maggior parte delle forme di Alzheimer risulta nel tipico esordio amnesico, mentre l’interessamento del tronco encefalico nel Parkinson è responsabile del disturbo del movimento. Con un percorso variabile, queste proteine si diffondono con un meccanismo prion-like, trans-sinaptico, innescando sostanzialmente sempre gli stessi processi già visti per le malattie prioniche: infiammazione e morte neuronale [10].
Si è detto che un decimo dell’APP è indirizzata alla via amiloidogenica. Se è vero che l’amiloide ha la capacità di diffondersi con il meccanismo sopra descritto, svilupperemo tutti l’Alzheimer? La risposta è “probabilmente no”, e la spiegazione dipende verosimilmente dalla biologia e dallo stile di vita. Dal punto di vista biologico, praticamente per ogni prodotto di scarto o nocivo prodotto dal nostro organismo esistono uno o più sistemi di smaltimento: questo meccanismo viene chiamato clearance, e l’amiloide cerebrale non ne è esente (anzi, esistono numerose vie attraverso cui l’amiloide viene smaltita). In particolare, il tasso di produzione e di clearance dell’Aβ è sostanzialmente uguale nel cervello sano, e pari a circa l’8%. I meccanismi di clearance comprendono per esempio lo smaltimento attraverso i vasi sanguigni ed il sistema glinfatico. Nel primo caso, l’Aβ passa dal tessuto cerebrale ai microvasi arteriosi cerebrali, venendo poi distrutta in periferia. Questo meccanismo è influenzato in parte da una proteina chiamata apolipoproteina E (APOE), un trasportatore del colesterolo che compete con l’Aβ per lo smaltimento attraverso il circolo. Diverse isoforme di APOE presentano una diversa capacità di mediare la clearance di Aβ, con la variante ε2 che risulta neuroprotettiva e la variante ε4 meno efficiente [11]. Sfortunatamente, l’APOE ε4 induce anche una maggiore produzione di Aβ [12]. Nel caso della clearance da parte del sistema glinfatico, l’Aβ passa nel liquido cefalorachidiano attraverso dei “tunnel” formati dagli astrociti, delle particolari cellule che si trovano tra neuroni e vasi, per poi passare nei vasi linfatici del collo e quindi nelle vene [13]. Questo meccanismo di clearance è influenzato dal sonno: infatti, mentre dormiamo (in particolare di lato) il sistema glinfatico è più attivo, mentre durante la veglia è sostanzialmente nullo [14]. Proprio le modificazione dei ritmi del sonno, potrebbero, in effetti, essere una delle prime spie delle alterazioni biologiche che sottendono la patologia di Alzheimer, in quanto il tratto retino-ipotalamico, che resetta ogni giorno l’orologio biologico cerebrale (il nucleo suprachiasmatico ipotalamico) portando le informazioni luminose marcatempo (il cosiddetto Zeitgeber), andrebbe incontro ad una degenerazione precoce, anche anni prima dello sviluppo dei sintomi cognitivi tipici della malattia [15].
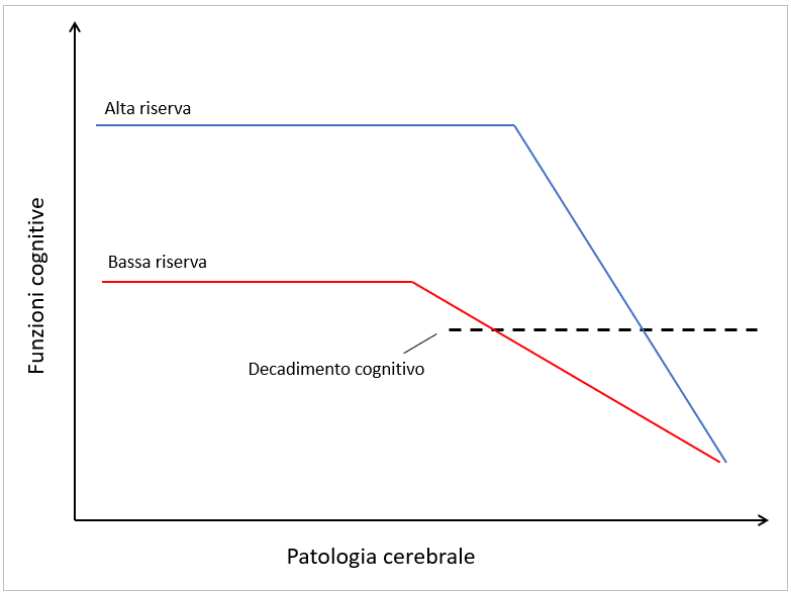
Dal punto di vista dello stile di vita, esistono dei meccanismi protettivi e dei meccanismi deleteri che influenzano la progressione biologica e l’espressione clinica della malattia. Quelli protettivi coincidono sostanzialmente con il concetto di “riserva cognitiva”. Con l’avanzare dell’età quasi tutti i meccanismi biologici di clearance tendono a fallire, per via della disfunzione di multipli sistemi, e in molti cervelli anziani l’accumulo di amiloide è significativo – tuttavia, non sempre questo comporta la presenza di decadimento cognitivo. Questo perché una riserva cognitiva più alta permetterebbe di sviluppare connessioni intracerebrali più efficienti e compensare un carico patologico maggiore. La riserva cognitiva è influenzata da vari fattori, tra cui la scolarità, l’attività cognitiva durante la vita (use it or lose it), la ricchezza del contesto sociale. Al contrario, una bassa scolarità e una vita povera di stimoli cognitivi renderebbero il cervello più indifeso contro l’accumulo di neuropatologia [16]. Tra i meccanismi deleteri troviamo invece l’inquinamento dell’aria [17], una dieta ricca di grassi, la scarsa attività fisica, il fumo, l’obesità, i traumi cranici e la depressione [18]. Per quanto riguarda l’alcool, il rischio relativo sembra dipendere dall’APOE: per i portatori di APOE ε4 è associato al rischio di Alzheimer, mentre per i non portatori quantità moderate sembrano essere protettive [19,20].
Fattori di rischio per lo sviluppo della malattia di Alzheimer
Da quanto detto fino ad ora, emergono due pattern con cui si può sviluppare la malattia di Alzheimer: un’aumentata produzione di Aβ, o una ridotta clearance. Al primo percorso appartengono i fattori di rischio genetici come le mutazioni del gene che codifica per la proteina APP, che si trova sul cromosoma 21 – è questo il motivo per cui gli individui affetti da sindrome di Down sviluppano la malattia di Alzheimer intorno ai 40 anni (la trisomia 21 infatti causa un’aumentata produzione di APP e quindi di Aβ). Le mutazioni di APP e delle preseniline (PSEN1 e PSEN2) possono inoltre influenzare il modo in cui l’APP viene clivata dalle secretasi, favorendo relativamente la via amiloidogenica. Queste tre mutazioni in particolare rappresentano il fattore di rischio genetico più elevato per lo sviluppo di Alzheimer in età giovanile (anche prima dei 50 anni), aumentando la probabilità di 15-20 volte, ma sono molto rare. L’APOE4, per contro, presenta un rischio di 3-4 volte superiore rispetto al normale, ma è molto più prevalente – un quarto della popolazione generale presenta almeno una copia su due di questa variante, che come detto agisce su due fronti: aumenta la produzione di amiloide, e ne riduce la clearance. Le forme associate ad APOE ε4 sono comunque più spesso ad esordio senile [21,22].
Al secondo percorso appartengono invece tutti quei fattori che riducono la capacità dell’organismo di smaltire una quantità sostanzialmente normale di amiloide. Tra questi, ve ne sono di non modificabili (come l’invecchiamento o la già citata APOE4) e di modificabili. Questi ultimi comprendono soprattutto aspetti già trattati, come il sonno, i traumi cranici e i fattori di rischio cardiovascolari.
Tuttavia, ci sono anche fattori protettivi: oltre ai già citati fattori che contribuiscono alla riserva cognitiva, alcune mutazioni conferiscono un rischio minore di sviluppare Alzheimer, come alcune varianti di APP (come la mutazione islandese, presente però in meno dell’1% degli islandesi), di APOE (come la variante Christchurch di APOE3) e della proteina reelina [23–25]. Per quanto, di per sé, tutte assai rare, lo studio di queste mutazioni protettive potrebbe fornire lo spunto per lo sviluppo di farmaci efficaci contro l’Alzheimer e agenti attraverso meccanismi completamente diversi rispetto a quelli finora perseguiti [26].
In generale, la forma più frequente di demenza di Alzheimer è quella senile, una forma mista con patologia di Alzheimer e aumentato carico vascolare, in cui una combinazione di fattori di rischio genetici a bassa penetranza e fattori legati allo stile di vita contribuiscono sinergicamente alla progressione del decadimento cognitivo [27].
Tra le domande che vengono più spesso poste in ambulatorio c’è quella sull’ereditarietà dell’Alzheimer. Vista la prevalenza enorme di questa malattia, non è infrequente che un adulto di mezza età abbia avuto almeno un parente affetto. L’ereditarietà è stimata intorno al 60%, ma questo è riferito soprattutto alla forma senile – che potenzialmente è anche quella più prevenibile, o almeno ritardabile, agendo sui fattori di rischio modificabili. Ovviamente, il rischio genetico cresce con il numero di parenti affetti; non è molto diverso da quello della popolazione generale quando si ha solo un parente affetto, ma può diventare significativo dopo i tre. Bisogna inoltre considerare che a prescindere dalla genetica, il rischio di sviluppare una demenza nell’arco della vita (ipotizzando uno span di 80 anni) è di circa il 24%, con circa la metà di questa percentuale dovuta ad Alzheimer [28,29].
Cosa si poteva fare, fino ad ora
Se per le forme familiari ad esordio precoce siamo ancora abbastanza lontani da soluzioni efficaci, sulle forme senili abbiamo invece un duplice armamentario terapeutico a disposizione. La prima categoria è quella dei farmaci “sintomatici”, che non rallentano la progressione biologica della malattia (o lo fanno solo in parte), ma possono dare un beneficio parziale su alcuni aspetti come la memoria e i disturbi del comportamento associati alla malattia di Alzheimer. Questi comprendono essenzialmente due categorie di farmaci: gli inibitori dell’acetilcolinesterasi e la Memantina.
La storia della prima classe raffigura un classico esempio di drug repurposing, che spesso si incontra in medicina, ovvero il riutilizzo di un farmaco utilizzato per tutt’altro scopo. La storia inizia nel 1947 con l’identificazione in territorio sovietico di un alcaloide ottenuto dal Galanthus nivalis, il comune bucaneve (nonché dal più spettacolare Lycoris radiata, o giglio del ragno rosso). Tale composto, chiamato Galantamina, venne all’inizio usato per trattare le paralisi post-polio in Bulgaria, Italia, Francia e Germania negli anni ’60, venendone successivamente ipotizzato un uso anche per la disfunzione erettile. Negli anni ’80 si ipotizzò che il meccanismo neurotrasmettitoriale alla base della demenza senile fosse legato ad una relativa progressiva perdita di acetilcolina cerebrale (analogamente a quanto era stato scoperto circa 15 anni prima per la dopamina nel Parkinson). L’acetilcolina viene usata dal nucleo basale di Meynert per “attivare” sostanzialmente quasi tutta la corteccia cerebrale. Non sorprendentemente, una delle prime aree cerebrali colpite dall’accumulo di amiloide nell’Alzheimer è proprio il nucleo basale di Meynert, il che riconcilia l’ipotesi colinergica e l’ipotesi dell’amiloide. L’idea dei ricercatori negli anni ’90 è stata quindi quella di somministrare dei composti che aumentassero l’acetilcolina nel cervello, tramite la somministrazione diretta di un suo precursore, la citicolina, oppure inibendo l’attività dell’enzima responsabile della sua degradazione, l’acetilcolinesterasi.
I primi studi riguardarono la Fisostigmina e la Tacrina, che venne poi ritirata per significativi effetti collaterali epatici. Seguirono il Donepezil, il Metrifonato, l’Uperzina, la Rivastigmina, tutti composti sintetici, con l’eccezione dell’Uperzina, estratta da una pianta asiatica chiamata Huperzia serrata. L’ultimo anticolinesterasico testato ed approvato per l’Alzheimer fu proprio la Galantamina, cioè il primo scoperto tra quelli disponibili [30]. Attualmente, gli anticolinesterasici approvati in Europa sono il Donepezil, la Rivastigmina (disponibile sia in compresse che nel più popolare cerotto) e la Galantamina, forse un po’ meno utilizzata. Sono indicati per le fasi lievi-moderate della malattia di Alzheimer (e della demenza a corpi di Lewy, un’altra malattia in cui è presente un significativo deficit colinergico). Sembra che solo i due terzi dei pazienti risponda a questi farmaci, e la risposta stessa è quantificabile nella maggior parte dei casi in una stabilizzazione dell’aspetto cognitivo per circa 3-9 mesi. Alcune caratteristiche possono aiutare a ipotizzare chi risponderà più probabilmente a questi farmaci, come la preservazione degli ippocampi, la presenza di allucinazioni e fluttuazioni cognitive, o una più veloce progressione di malattia prima dell’inizio della terapia [31].

La Memantina invece basa il suo effetto sul blocco della trasmissione glutammatergica. Il glutammato è emerso come un bersaglio molto interessante nelle malattie neurodegenerative dopo l’osservazione che il consumo dei semi di Cycas micronesica sull’isola di Guam, e del pisello Lathyrus sativus in generale nel mondo, portava a due quadri molto simili alla sclerosi laterale amiotrofica (la SLA). In particolare, la forma di Guam era una malattia che colpiva i Chamorro, chiamata Lytigo-bodig o ALS-Parkinson-Dementia complex, una coesistenza variabile di sclerosi laterale amiotrofica, demenza con alterazioni del comportamento e difficoltà linguistiche (forma lytico), Parkinson (forma bodig), e alterazioni patologiche tipiche. Tutto insieme. Il neurologo Oliver Sacks, ne “L’isola dei senza colore”, un libro bellissimo che parte dai Kew Gardens per arrivare all’atollo micronesico di Pingelap e a Guam, riporta le sue osservazioni sul campo in merito alla malattia. Per ciò che si sa attualmente, i Chamorro utilizzavano i semi di cycas per preparare una farina chiamata fadang, con l’effetto collaterale di sviluppare la malattia. Tuttavia, l’interruzione del consumo di cycas non aveva del tutto eliminato la malattia. Ciò è stato ipotizzato essere dovuto al fatto che i Chamorro continuavano a nutrirsi di pipistrelli, che a loro volta si nutrivano di cycas, bioconcentrando la tossina responsabile del quadro (analogamente a quanto avviene per il mercurio nel pesci man mano che ci si muove verso l’alto nella catena alimentare) [32,33].
Il neurolatirismo è invece una malattia legata al consumo di semi cotti di cicerchia (il Lathyrus sativus), alla base di piatti tipici europei come le gachas manchegas in Spagna. Le popolazioni umane hanno consumato questo alimento soprattutto durante i periodi di carestia dovuta a siccità, perché la cicerchia cresce più facilmente di altri vegetali; per contrastarne la tossicità, la bollitura dei semi è stata utilizzata nei secoli come rimedio relativamente semplice. Tuttavia, la siccità è responsabile della minore disponibilità di acqua per bollire i semi di cicerchia, oltre che del suo maggiore consumo. Questi fattori combinati hanno portato alla maggiore ingestione della tossina in essa contenuta, con l’emergenza di una malattia simile alla SLA, chiamata appunto neurolatirismo, con paralisi delle gambe, atrofia dei glutei e a volte anche problemi a carico di vasi sanguigni e ossa.

Ciò che accomuna la cycas e la cicerchia è la presenza di una tossina (in realtà due tossine diverse) che rappresenta un analogo del glutammato, il principale neurotrasmettitore eccitatorio del cervello, fondamentale per la memoria, il movimento e altre funzioni. L’eccesso di glutammato è tossico per i neuroni, in un meccanismo che viene chiamato “eccitotossicità” [34].
Questo meccanismo è implicato anche nella malattia di Alzheimer, sebbene non sia probabilmente il meccanismo centrale: infatti, l’accumulo di amiloide e l’eccitotossicità da glutammato si alimentano a vicenda, amplificando il danno sui neuroni. Sulla base di queste osservazioni, si è ipotizzato il glutammato come bersaglio potenziale per le malattie neurodegenerative, il che ha portato all’introduzione del Riluzolo nella SLA, in cui prolunga di qualche mese la sopravvivenza, e della Memantina nell’Alzheimer, in cui dà un lieve beneficio sintomatico [35]. Quest’ultimo farmaco viene in genere utilizzata nelle fasi moderate della demenza, e può dare anche effetti positivi sull’agitazione nei pazienti con Alzheimer [36]. Tuttavia, pur combinando gli inibitori dell’acetilcolinesterasi e la Memantina, il trattamento dell’Alzheimer è stato fino ad oggi decisamente poco soddisfacente.
Cosa si può fare, da oggi
Il Sacro Graal per chi si occupa di malattie neurodegenerative è dato dalle terapie disease-modifying, cioè da quei trattamenti agenti sulla causa sottostante alla malattia e in grado di cambiare significativamente il decorso della malattia. In questo sono sostanzialmente diversi dai farmaci sintomatici, che invece trattano solo il prodotto della malattia, ovvero, appunto, i sintomi [37]. Negli ultimi vent’anni sono stati fatti sostanziali sforzi alla ricerca di trattamenti disease-modifying, principalmente rivolti verso l’amiloide e la tau. I trattamenti più promettenti sono sembrati quelli con anticorpi monoclonali volti a “ripulire” il cervello dagli aggregati di Aβ, sostanzialmente facilitandone la clearance (gli inibitori della β-secretasi sono invece falliti [38]).
Il primo anticorpo monoclonale ad essere approvato dall’FDA nel 2021 è stato l’Aducanumab, prodotto dalla Biogen con il nome commerciale di Aduhelm. La storia di questo farmaco è molto confusa, e non scevra di polemiche, culminate nella pirotecnica dimissione di tre membri dell’Advisory committee dell’FDA e nel ritiro della richiesta di approvazione del farmaco in Europa dopo una prima sostanziale bocciatura (https://www.ema.europa.eu/en/medicines/human/withdrawn-applications/aduhelm) [39,40]. In breve, due studi randomizzati paralleli di fase III (l’ultima prima dell’approvazione di un farmaco) di Aducanumab contro placebo nelle fasi iniziali dell’Alzheimer, EMERGE ed ENGAGE, sono stati interrotti precocemente per un’analisi di futilità. Tuttavia, una successiva sotto-analisi sembrava suggerire che la dose più alta di farmaco, da 10 mg/kg (che viene raggiunta solo dopo alcuni mesi), potesse conferire un beneficio clinico rallentando – di poco – la malattia e riducendo l’amiloide nel cervello, come si poteva vedere dall’analisi delle PET con tracciante per amiloide. L’approvazione accelerata da parte dell’FDA è stata basata sul raggiungimento di un endpoint surrogato (cioè l’effetto biologico di riduzione dell’amiloide cerebrale), e non sull’obiettivo primario (cioè l’efficacia clinica, che non è stata raggiunta), il che rappresenta un’anomalia notevole nel processo di approvazione dei farmaci. A Biogen sono stati dati nove anni per dimostrare l’efficacia clinica del farmaco in un nuovo trial, EMBARK, tutt’ora in corso (https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/aducanumab-marketed-aduhelm-information). Verrebbe da chiedersi se per una malattia così devastante quel poco che potrebbe fare Aducanumab non sia comunque meglio del pochissimo che abbiamo al momento, e questa è spesso una delle motivazioni che spingono il pubblico a parteggiare per l’immissione in commercio di farmaci di non provata efficacia. Tuttavia, il problema è chiaramente molto più complesso, principalmente per due ragioni: gli effetti collaterali e i costi.
Gli effetti collaterali più seri di questa terapia sono le cosiddette amyloid-related imaging abnormalities (ARIA), che possono essere di due tipi: ARIA-E, con edema cerebrale (cioè accumulo di liquidi dovuto ad infiammazione), o ARIA-H, con emorragia cerebrale. Le ARIA ricordano molto da vicino due aspetti di una malattia chiamata angiopatia amiloide cerebrale (o CAA), in cui l’Aβ si deposita all’interno dei piccoli vasi invece che nel tessuto cerebrale, causandone la rottura con sanguinamento, l’occlusione con ictus, e in alcuni casi infiammazione da anticorpi contro l’Aβ (nella CAA-RI, o CAA-related inflammation). Anche questa malattia è legata all’alterata clearance dell’Aβ, e può presentarsi insieme alla malattia di Alzheimer [41]. Per le ARIA il meccanismo sembra essere simile – le ARIA-E sarebbero legate all’infiammazione e alla saturazione dei meccanismi di clearance (con una maggiore associazione con l’APOE4), mentre le ARIA-H dipenderebbero dalla clearance dell’amiloide vascolare con un meccanismo simile a ciò che avviene nella CAA (ed associazione con età e malattie cerebrovascolari) [42]. Negli studi iniziali sull’Aducanumab, il 40% dei pazienti trattati con la dose da 10 mg/kg (quella ritenuta potenzialmente efficace) ha sviluppato ARIA. Il 35% ha sviluppato ARIA-E, sintomatica in un caso su quattro (con cefalea, confusione, vertigini, ecc), mentre il 20% ha sviluppato ARIA-H (con microsanguinamenti). Anche se la maggior parte dei pazienti aveva avuto eventi lievi, alcuni pazienti avevano avuto sintomi anche gravi [43]. Ciò comporta due conseguenze. La prima è che se le ARIA possono essere anche asintomatiche, è necessario sottoporre i pazienti a risonanze magnetiche molto frequentemente (almeno quattro in un anno), per valutare se il trattamento sta creando problemi e decidere se e come proseguire. La seconda è che non tutti i pazienti sono candidabili per il trattamento con Aducanumab, e per decidere quali pazienti possono beneficiare della terapia o rischiare di avere eventi avversi sono necessari biomarker costosi e l’analisi genetica dell’APOE [44]. Nel mondo reale, sempre un po’ meno emozionante di quello dei trial, l’eleggibilità per l’Aducanumab si attesterebbe al massimo ad un paziente su dieci [45].
Per quanto riguarda i costi, alcune analisi di cost-effectiveness hanno riportato che forse il gioco non vale la candela. Il prezzo iniziale dell’Aducanumab era di 56.000 dollari all’anno, giudicato decisamente troppo alto comparato con il risparmio atteso (cioè la riduzione dei costi correlati alla progressione della demenza, anche nell’improbabile caso in cui l’Aducanumab bloccasse completamente la progressione della malattia). Poco dopo l’approvazione di Aducanumab, Biogen ha annunciato una riduzione del prezzo di circa la metà, ma nemmeno in questo caso passerebbe l’analisi costo-efficacia (sarebbe necessario un prezzo sotto i 22.000 dollari) [46]. Questo senza considerare l’impreparazione nell’affrontare non solo i costi, ma anche la logistica che queste terapie implicano: posti in risonanza, disponibilità di biomarkers per la selezione dei pazienti, luoghi in cui effettuare queste terapie (che sono endovena), personale implicato. Nessun sistema sanitario al mondo è attualmente preparato per l’implementazione di queste terapie [47,48].
Sulla scia di quanto avvenuto con Aducanumab, a metà di quest’anno è stato approvato negli Stati Uniti il Lecanemab, un altro anticorpo monoclonale contro Aβ. Il meccanismo è stato ancora una volta quello dell’approvazione accelerata (cioè basata su un marker surrogato – la riduzione della beta-amiloide – piuttosto che sull’evidenza di un reale beneficio clinico – il rallentamento della progressione dei deficit cognitivi). Dal 6 luglio 2023 la FDA ha fornito la piena approvazione al Lecanemab (https://www.fda.gov/news-events/press-announcements/fda-converts-novel-alzheimers-disease-treatment-traditional-approval). Il Lecanemab sembra in realtà essere più potente dell’aducanumab, ed anche più efficace: la riduzione di amiloide cerebrale è maggiore, e sembrano esserci effetti significativi sulla progressione del decadimento cognitivo, sulla qualità di vita, sull’indipendenza funzionale dei pazienti nelle prime fasi della demenza [49]. La deposizione di amiloide cerebrale può essere quantificata con la PET-amiloide con una particolare unità di misura chiamata centiloide [50]. Il limite per considerare una PET positiva è di 30 centiloidi, e negli studi si è valutata sia la riduzione dei centiloidi dal baseline, sia il raggiungimento di un valore sotto soglia. Dai risultati degli studi, sia Aducanumab, che Lecanemab, che Gantenerumab (un anticorpo monoclonale della Roche) causavano una riduzione di circa 50-60 centiloidi. Questo tuttavia è avvenuto in modo diverso nei due studi dell’Aducanumab: in EMERGE, lo studio in cui si è osservata un’efficacia clinica relativa, la riduzione media nel gruppo che ha raggiunto la dose di 10 mg/kg era di 64 centiloidi, con una media finale di circa 25 centiloidi (negativizzando quindi la PET-amiloide), mentre in ENGAGE, che non ha raggiunto l’endpoint primario, la riduzione era di 53 centiloidi, con una media finale di circa 37 (ancora positiva) [51]. Nello studio CLARITY-AD sul Lecanemab, la riduzione media è stata di 59 centiloidi, con una media finale sotto 30 centiloidi [49]. Nei due studi su Gantenerumab, GRADUATE I e II, la riduzione è stata tra i 48 e i 57 centiloidi, ma solo un quarto dei pazienti ha ottenuto un valore sotto soglia alla fine del trial, che non ha raggiunto neanche il suo endpoint clinico primario [52]. La Roche ha quindi chiuso gli studi in corso, compreso il POST-GRADUATE (che estendeva i precedenti), alzando bandiera bianca su un percorso iniziato addirittura vent’anni prima. L’insegnamento che si può trarre da questi studi è che verosimilmente ripulire il cervello dall’amiloide in maniera pressoché completa dà risultati clinici reali.
È probabile che questa maggiore efficacia, con peraltro meno ARIA (circa il 10-15%, quasi nulle tra i non portatori di APOE4), abbia in parte a che vedere con l’effetto di una diversa selettività per il tipo di Aβ. Il Lecanemab infatti è più selettivo contro gli oligomeri e le protofibrille solubili, agendo in un punto del processo più a monte rispetto all’Aducanumab, che è più selettivo per le fibrille insolubili [53,54].

Del tutto da valutare è però l’impatto clinico reale del “successo” dei trial – l’obiettivo primario per tutti è la CDR-Sum of Boxes, una scala da 0 a 18 che valuta l’impatto della malattia su sei domini, tra cui la memoria, la capacità di esprimere un giudizio, la cura personale, l’orientamento. I cut-off generalmente usati prevedono ampi margini per passare da una categoria di severità alla successiva.
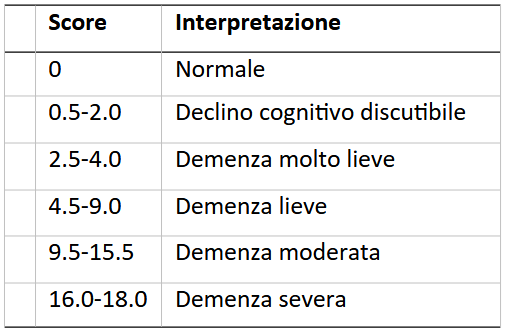
Persino nel trial più efficace, la differenza tra Lecanemab e placebo, per quanto significativa statisticamente, non lo era probabilmente dal punto di vista clinico, riducendosi a 0.4 punti di media a diciotto mesi (ovvero ad un 27% di rallentamento della malattia). I pazienti perdevano 1.2 punti con il farmaco, e 1.6 con il placebo. Ma cosa significano quindi questi 0.4 punti? Sostanzialmente niente. Un paziente potrebbe partire da 2.5, una demenza molto lieve, e raggiungere 3.5 e 4.0 con il Lecanemab e senza, ricadendo sostanzialmente ancora nella stessa categoria. A “breve termine” è molto difficile che l’impatto di questi farmaci sia importante, mentre a lungo termine possiamo per il momento solo fare supposizioni.
Lilly ha annunciato effetti molto positivi per il suo monoclonale, il Donanemab, diretto principalmente contro le placche di amiloide. I risultati completi verranno presentati in occasione del congresso sull’Alzheimer in programma ad Amsterdam il prossimo luglio (https://aaic.alz.org/), ma nel frattempo viene riportato un rallentamento del 37% alla CDR-Sum of Boxes (che tuttavia non era l’outcome primario), ed una riduzione anche sull’accumulo di tau, la seconda proteina ad accumularsi nell’Alzheimer. Proprio contro tau i farmaci fino ad ora provati si sono rilevati un fallimento, anche se qualche dato incoraggiante si inizia a profilare all’orizzonte [55,56].
Sia per il Lecanemab che per il Donanemab rimangono aperte le questioni di ARIA [57] e costi (con il prezzo che si aggira sui 26.000 dollari all’anno, e ancora non cost-effective [58,59]), oltre che quella dell’eleggibilità. I costi in realtà potrebbero essere ancora più alti, e fino a 90.000 dollari l’anno includendo tutte le risorse necessarie per la prescrizione ed il monitoraggio della terapia, come risonanze, PET, genetica, strutture per le infusioni e per la gestione delle complicanze – negli Stati Uniti, nonostante la copertura da parte di Medicare, ai pazienti potrà essere richiesto di pagare il 20% di questo costo totale, il che non è chiaramente sostenibile per tutti [60–62]. E non per tutti potrebbe valerne la pena. In particolare, analizzando i dati ad oggi a disposizione dal trial CLARITY-AD sul Lecanemab, emerge come il candidato ideale sia un uomo bianco sopra i 65 anni con al massimo una copia di APOE4 su due. Nelle donne, nelle persone di etnia asiatica o di colore (estremamente sottorappresentate nello studio e nella ricerca in generale), nelle forme di Alzheimer early-onset (prima dei 65 anni) e negli omozigoti per APOE4 il farmaco non ha mostrato differenze significative rispetto al placebo [63]. Resta dunque da chiarire quale sia l’utilità reale di un farmaco che costa moltissimo, può dare effetti collaterali anche gravi, non modifica di molto la traiettoria di malattia ed è efficace solo in alcuni soggetti – nonostante queste perplessità, si tratta comunque di un passo in avanti nella terapia, il più grande passo da vent’anni a questa parte. Eisai, l’azienda produttrice di Lecanemab, ipotizza che l’uso continuativo di Lecanemab dalle primissime fasi di decadimento cognitivo (quando il paziente è ancora sostanzialmente autonomo) possa ritardare di anni la progressione alla fase severa di demenza, ma bisognerà attendere dei dati real-world per valutare l’effettivo impatto della terapia.
Cosa si potrà fare, da domani
Nel frattempo, Eisai si è già mossa in anticipo, lanciando nel 2020 due trial paralleli, AHEAD-3 e AHEAD-45, in cui il Lecanemab viene somministrato per 4 anni a soggetti tra 55 e 80 anni senza deficit cognitivi, ma con evidenza di aumentata deposizione di amiloide cerebrale (rispettivamente 20-40 e più di 40 centiloidi) [64]. Questi soggetti, secondo i criteri di ricerca più aggiornati, hanno già la malattia di Alzheimer dal punto di vista biologico, ma non l’hanno ancora sviluppata nella sua espressione clinica – ovvero il decadimento cognitivo [65]. A dire il vero, questi soggetti possono anche manifestare delle alterazioni cognitive soggettive, ma i test neuropsicologici standardizzati non risulteranno diversi da ciò che è atteso in base all’età. Torneremo più avanti su questa popolazione. In ogni caso, con i trial AHEAD si sta provando per la prima volta a prevenire l’Alzheimer con un farmaco, cosa che fino ad una decina di anni fa sembrava pura fantascienza; vedremo se lo studio darà i risultati sperati.
È tuttavia probabile che anche in questo caso qualcuno rimarrà escluso – e i dati riportati fino ad ora fanno pensare che siano i portatori di APOE4. Ma cosa si può fare in questi soggetti? Premesso che essere portatore di APOE4 non conferisce la certezza di sviluppare l’Alzheimer, ma solo un rischio aumentato, questo gruppo sembra essere quello che più può beneficiare dall’implementazione di interventi preventivi multidimensionali sullo stile di vita [66].
È concezione diffusa, probabilmente derivante da un’interpretazione sbagliata di una citazione di Giovenale, che nel mondo greco-romano la salute del corpo fosse una conditio sine qua non per la salute mentale. In realtà, la frase che compare nella satira X del poeta romano non è la famosa “mens sana in corpore sano”, bensì “orandum est ut sit mens sana in corpore sano”, che più che la moderna accezione del detto, significa “bisogna pregare di avere una mente sana in un corpo sano”. Se volessimo riassumere il contenuto della satira di Giovenale con un proverbio moderno, potremmo usare più convenientemente “quando c’è la salute c’è tutto” o “chi ha la salute è ricco e non lo sa”. Nell’età classica era invece più diffusa la credenza che l’attività fisica fosse in qualche modo in antitesi con l’attività mentale, o quanto meno che l’una necessitasse di essere temperata dall’altra.
A prescindere dalle questioni storiche, più o meno negli stessi anni in cui si sono svolti i trial sui farmaci anti-amiloide ci si è resi conto che intervenire sui fattori di rischio che abbiamo esposto nei capitoli precedenti poteva essere una strategia efficace nella prevenzione della demenza. La vera differenza rispetto a quanto fatto prima riguardava l’approccio, che in precedenza era stato monodimensionale, valutando l’impatto di un singolo intervento alla volta – come attività fisica, stimolazione cognitiva, dieta. In genere questi studi non davano risultati entusiasmanti, probabilmente perché i meccanismi implicati nella neurodegenerazione e nel decadimento cognitivo sono complessi, e concentrarsi su un solo fattore è troppo poco. A dire il vero, non tutti gli studi multidimensionali hanno dato i risultati sperati.
I tre trial più importanti in questo senso sono stati il FINGER, il MAPT e il Pre-DIVA. Né il MAPT né il Pre-DIVA hanno mostrato un’efficacia degli interventi multidimensionali nel prevenire la demenza; entrambi gli studi arruolavano soggetti di almeno 70 anni, nel primo caso con almeno uno di tre fattori di rischio (deficit di memoria soggettivi, limitazioni nelle attività della vita quotidiana, o cammino lento), e nel secondo senza particolari criteri di selezione aggiuntivi. Il FINGER al contrario interessava una popolazione più giovane (60-77 anni), prevedeva cinque domini di intervento (attività fisica, dieta, controllo dei fattori di rischio cardiovascolare e metabolico, stimolazione cognitiva e socializzazione), e tra i criteri di selezione c’era la presenza di un punteggio di 6 o più al CAIDE – una scala che può predire lo sviluppo di demenza nei successivi 10-20 anni [67]. Il FINGER prevedeva inoltre una maggiore personalizzazione degli interventi rispetto agli altri due trial, ed effettivamente è stato l’unico a raggiungere l’endpoint primario, con un follow-up che attualmente è stato programmato fino ad undici anni per valutarne gli effetti a lungo termine. Tuttavia, delle sottoanalisi hanno mostrato che anche l’approccio degli altri trial era efficace in gruppi a maggior rischio di sviluppare demenza [68,69]. Il metodo FINGER si basa fondamentalmente su tre principi: gli interventi devono essere multidimensionali, personalizzati, e applicati nel giusto momento. Infatti, per specifici fattori di rischio ci sono finestre temporali diverse, per cui per esempio il trattamento dell’ipertensione ha un’importanza relativa maggiore in mezza età piuttosto che nell’età senile [18]. Sulla base dell’esperienza accumulata in questi studi, è nato un vero e proprio network, il FINGER WORLD-WIDE, con l’intento di avviare più studi adattati allo specifico contesto culturale di diversi paesi, tra cui il FINGER-NL nei Paesi Bassi, il POINTER negli Stati Uniti, il PENSA in Spagna. Questi studi differiscono tra loro nel tipo di domini di intervento, con qualcuno che inserisce per esempio il miglioramento del sonno o dello stress, o l’utilizzo di integratori, e nei criteri di selezione dei pazienti, per esempio includendo soggetti a rischio genetico aumentato, o portatori di APOE4, o affetti da decadimento cognitivo soggettivo [70]. Per quanto riguarda i singoli interventi, il programma è abbastanza vario. Per la dieta si tende a proporre la dieta mediterranea, o una sua variante (la dieta MIND, che sembra ridurre il rischio di progressione dell’Alzheimer) [71,72]. Per l’attività fisica invece viene solitamente offerta una combinazione di attività aerobica e allenamento in palestra, con frequenza variabile. L’implementazione dei restanti interventi è ancora più varia, con alcuni studi che prevedono di fare più attività possibili in gruppo per promuovere la socializzazione.
Anche in Italia inizia a muoversi qualcosa in tal senso. È stato recentemente approvato nell’ambito del Programma Nazionale di Ripresa e Resilienza (PNRR) un progetto chiamato “Age-It: conseguenze e sfide dell’invecchiamento”, coinvolgente numerose università italiane, volto a trasformare l’Italia in un hub di ricerca sull’invecchiamento ed in particolare sull’invecchiamento in salute. Nell’ambito di questo mega-progetto alcuni spoke, tra cui quello in cui chi scrive si trova a lavorare, sono dedicati proprio all’implementazione di interventi per ridurre il carico delle malattie correlate all’età (https://ageit.it/vp-3-temi-di-ricerca.html). È ovvio che questo include anche la malattia di Alzheimer. Uno dei sotto-progetti di questo spoke sarà proprio volto a valutare il potenziale preventivo di interventi multidimensionali ispirati al trial FINGER sul decadimento cognitivo e funzionale, agendo su determinate popolazioni a rischio e sfruttando l’impatto delle nuove tecnologie.
Quando agire: i Brain Health Services
La spinta verso la prevenzione dell’ultimo decennio ha portato alla definizione di una nuova categoria “diagnostica”, comprendente soggetti sani ad aumentato rischio di sviluppare decadimento cognitivo per via delle loro caratteristiche cliniche. Si tratta dei soggetti affetti da Decadimento Cognitivo Soggettivo (Subjective Cognitive Decline, SCD), i cui criteri sono stati definiti nel 2014 ed integrati negli anni successivi. La definizione prevede una serie di condizioni:
- Declino persistente e soggettivo nelle capacità cognitive rispetto ad uno stato precedente normale, non collegato ad un evento acuto
- Performance normale per età, sesso ed istruzione nei test cognitivi standardizzati utilizzati per diagnosticare il decadimento cognitivo lieve o l’Alzheimer prodromico
I criteri di esclusione comprendono la presenza di decadimento cognitivo oggettivabile, o la possibilità che i disturbi siano spiegabili da una condizione psichiatrica (come una depressione grave), neurologica (ad esclusione dell’Alzheimer), medica, o da un abuso di sostanze. Dalla definizione seguono tre concetti fondamentali: che l’SCD non è una forma precoce di Alzheimer, ma un “campanello d’allarme”, che l’SCD non è legato ad eventi acuti (come un lutto familiare, o un periodo particolarmente stressante), e che aspetti depressivi lievi non escludono la definizione di SCD [73]. In sostanza, è normale sentire di “funzionare” peggio in situazioni che comportano un importante carico emotivo, ed un certo grado di depressione reattiva può accompagnare l’esperienza di difficoltà cognitive.
Oggi si tende a distinguere la malattia di Alzheimer biologica (con la positività di biomarkers per beta-amiloide, tau e neurodegenerazione) dalla malattia di Alzheimer clinica (con decadimento cognitivo). L’SCD può rappresentare una fase di Alzheimer “preclinica” solo in quei casi in cui c’è una positività dei marcatori potendo essere peraltro dovuta a molte altre condizioni neurodegenerative e non. Negli ultimi anni sono stati proposti ed affinati dei criteri che aumentano la possibilità che l’SCD sia dovuto ad una sottostante malattia di Alzheimer, aumentando quindi il rischio di progressione a decadimento cognitivo oggettivo. Questa condizione è detta “SCD-plus”, e singoli aspetti presentano un rischio diverso di progressione e di riflettere una condizione di Alzheimer preclinico [74–76]. I soggetti che rispondono a questi criteri sono quelli in cui avrà probabilmente più senso proporre la ricerca dei marcatori di malattia di Alzheimer.

L’impatto dell’SCD non è banale. Da studi internazionali la condizione sembra interessare circa un quinto dei soggetti sopra i 45 anni, ed un quarto sopra i 60; la prevalenza è maggiore tra gli uomini, nei paesi del secondo e terzo mondo, negli asiatici e negli africani, e nelle persone con ridotti livelli di istruzione [77,78]. Incidentalmente, i fattori di rischio per la demenza, e quindi le possibilità e strategie preventive, variano in base alla regione del mondo [79]. I dati attualmente disponibili derivano da un certo numero di studi longitudinali sull’invecchiamento facenti parti di un consorzio mondiale (https://cheba.unsw.edu.au/consortia/cosmic/studies), oltre che da alcune coorti che arruolano esclusivamente soggetti con SCD (come la coorte SCIENCe olandese). Queste ultime iniziative sono attualmente ancora sporadiche e non standardizzate, e limitate a grossi centri con notevole expertise.
Due anni fa i principali ricercatori nel campo delle demenze hanno prodotto un “manuale” per un nuovo tipo di servizio rivolto alle fasi precliniche del decadimento cognitivo, con l’obiettivo di salvaguardare la salute cerebrale. Il nome proposto per queste nuove realtà è stato quello di Brain Health Services (BHS). Gli utilizzatori dei BHS sono individuati sostanzialmente in tre categorie di persone anziane e di mezza età: i soggetti con SCD, i soggetti con disturbi cognitivi funzionali (cioè con disturbi cognitivi nel contesto di ansia, depressione lieve e dolore cronico – sarebbero esclusi i pazienti con disturbi psichiatrici e dell’umore gravi, più efficacemente riferiti agli psichiatri), e i “worried wells”, cioè quelle persone senza disturbi, ma preoccupate per un eventuale futuro declino cognitivo, spesso influenzati dalla presenza di familiari affetti da demenza. Questi soggetti verranno in una prima fase riferiti ai BHS dai tradizionali ambulatori per i disturbi cognitivi (in Italia denominati Centri per i Disturbi Cognitivi e le Demenze o CDCD), ma in una successiva fase l’accesso sarà più verosimilmente spontaneo, e favorito dall’implementazione di campagne educative.
La funzione dei BHS sarà articolata in cinque fasi. La prima sarà profilare il rischio di demenza individuale, ricercando eventuali eventi acuti causanti il quadro di presentazione (a cui seguirà sostanzialmente l’interruzione del percorso e la rassicurazione del soggetto), la ricerca di fattori di rischio modificabili e non modificabili, l’utilizzo di score come il CAIDE, il Brief Dementia Screening Indicator o l’Australian National University AD Risk Index, l’utilizzo di biomarkers in soggetti selezionati (e per ricerca), ed eventualmente calcolatori di rischio [80]. A questa fase seguirà la comunicazione del rischio, con l’aiuto di psicologi e seguendo protocolli comunicativi validati. Successivamente, ai soggetti così inquadrati verranno proposti interventi preventivi di riduzione del rischio (o nel caso di soggetti con disturbi funzionali, un percorso specifico dedicato basato sulla spiegazione del quadro: https://neurosymptoms.org/it_IT/symptoms/fnd-symptoms/functional-cognitive-symptoms/). Questi interventi preventivi sono sostanzialmente quelli già individuati nell’ambito dei trial gemmati dallo studio FINGER, e basati sulla personalizzazione di interventi basati sulle caratteristiche individuali grazie ad un team multidisciplinare comprendente dietologi, psicologi, fisiologi dello sport, ed altre figure. In una fase ancora ulteriore, ai soggetti potrebbe essere proposto anche l’enhancement cognitivo, con training mentale o stimolazione magnetica transcranica (TMS), che al netto della necessità di maggiori studi sembra essere promettente. Infine, l’ultimo obiettivo dei BHS sarà la ricerca sulle condizioni precliniche dei disturbi neurodegenerativi, in una sorta di do ut des: ai soggetti viene offerto un percorso di definizione e riduzione del rischio sulla base delle conoscenze attuali, e gli stessi soggetti forniscono i loro dati clinici e biologici per l’aumento di queste conoscenze stesse. Chiaramente, a queste persone viene offerta anche la possibilità di rientrare nel percorso delle Memory Clinics o CDCD alle prime avvisaglie di una progressione verso il decadimento cognitivo lieve oggettivo, in modo che siano indirizzabili alle eventuali terapie farmacologiche delineate più sopra [81–86]. Tutto ciò rientra nel più ampio progetto della Brain Health Strategy, con cui la Neurologia europea sta cercando di modificare lo scenario socio-sanitario per promuovere la salute del cervello (https://www.ean.org/ean/advocacy/brain-health/brain-health-strategy).

Un’iniziativa di questo genere è stata avviata a giugno 2023 anche presso la Fondazione IRCCS San Gerardo dei Tintori (Monza), e per quanto di nostra conoscenza dovrebbe essere tra le prime realtà italiane in questo senso. La denominazione proposta è “ambulatorio SCD”, per il momento inquadrato all’interno del CDCD dell’Ospedale, con l’obiettivo di porre le basi per la successiva trasformazione in BHS secondo gli ultimi indirizzi proposti in letteratura, offrendo stima del rischio ed interventi preventivi [87]. L’accesso sarà per ora in base alla segnalazione attraverso il CDCD e gli ambulatori di Neurologia generale, ma in futuro si prevederà la possibilità di riferire i soggetti da parte dei medici di medicina generale opportunamente formati, e probabilmente anche attraverso l’interesse personale suscitato dalla pubblicizzazione delle attività del centro tramite eventi e simposi.
È cambiato moltissimo da quella prima conferenza a Tübingen in cui Alois Alzheimer è stato ignorato. Dai primi studi sulle alterazioni neuropatologiche associate alla demenza siamo giunti a proporre i primi farmaci che potrebbero funzionare, e una serie di interventi che potrebbero prevenire il decadimento cognitivo in soggetti che prima tendevamo solo a rassicurare. Dalla ricerca di caratteristiche comuni per spiegare un fenomeno complesso come la malattia di Alzheimer, stiamo correndo verso la medicina di precisione, per dare il giusto trattamento, alla giusta dose, alla giusta persona, al giusto momento. E prevedendo che questo trattamento, più che un rimedio tardivo ad una condizione già patologica, possa essere un modo per ritardare la malattia più a lungo possibile, preservando la salute del cervello. Perché, come diceva già più di trecento anni fa un medico di Carpi, prevenire è meglio che curare.
Ringraziamenti
Questo articolo è stato realizzato nel contesto del cofinanziamento dell’Unione europea –Next Generation EU, nell’ambito del Piano Nazionale di Ripresa e Resilienza (PNRR), Investimento Partenariato Esteso PE8 “Conseguenze e sfide dell’invecchiamento”, Progetto Age-It (Ageing Well in an Ageing Society).
Federico Emanuele Pozzi
Ildebrando Apollonio
Carlo Ferrarese
Lucio Tremolizzo
Clinica Neurologica, Fondazione IRCCS “San Gerardo dei Tintori”, Monza
Scuola di Medicina e Chirurgia, Università di Milano-Bicocca
Centro di Neuroscienze di Milano (NeuroMI), Università di Milano-Bicocca
Corrispondenza:
Dott. Federico Emanuele Pozzi
E-mail: federicoemanuele.pozzi@gmail.com
References
- Franco G. The 1711 rinderpest in bernardino ramazzini’s xiii oration and the covid-19 public health emergency: Facts and common aspects. Med del Lav. 2020;111(4):321–5. doi: 10.23749/mdl.v111i4.9672
- Lucci B. The contribution of Gaetano Perusini to the definition of Alzheimer’s disease. Ital J Neurol Sci. 1998;19(1):49–52. doi: 10.1007/BF03028813
- C. C. Peng F. Is alzheimer’s disease a fiction? Clin Res Trials. 2016;2(1):106–9. doi: 10.15761/crt.1000125
- Nichols E, Steinmetz JD, Vollset SE, Fukutaki K, Chalek J, Abd-Allah F, et al. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet Public Heal. 2022;7(2):e105–25. doi: 10.1016/S2468-2667(21)00249-8
- Zhang YW, Thompson R, Zhang H, Xu H. APP processing in Alzheimer’s disease. Mol Brain. 2011;4(1):1–13. doi: 10.1186/1756-6606-4-3
- Murphy MP, Levine H. Alzheimer’s Disease and the Amyloid-β Peptide. J Alzheimers Dis [Internet]. 2010;19:311–23. doi: 10.3233/JAD-2009-1221
- Bloom GS. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014;71(4):505–8. doi: 10.1001/jamaneurol.2013.5847
- Simonsen AH, Herukka SK, Andreasen N, Baldeiras I, Bjerke M, Blennow K, et al. Recommendations for CSF AD biomarkers in the diagnostic evaluation of dementia. Alzheimer’s Dement. 2017;13(3):274–84. doi: 10.1016/j.jalz.2016.09.008
- Engelborghs S, Niemantsverdriet E, Struyfs H, Blennow K, Brouns R, Comabella M, et al. Consensus guidelines for lumbar puncture in patients with neurological diseases. Alzheimer’s Dement Diagnosis, Assess Dis Monit [Internet]. 2017;8:111–26. doi: 10.1016/j.dadm.2017.04.007
- Jaunmuktane Z, Brandner S. Invited Review: The role of prion-like mechanisms in neurodegenerative diseases. Neuropathol Appl Neurobiol. 2020;46(6):522–45. doi: 10.1111/nan.12592
- Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, et al. Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol. 2015 Aug;11(8):457–70. doi: 10.1038/nrneurol.2015.119
- Hampel H, Hardy J, Blennow K, Chen C, Perry G, Kim SH, et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol Psychiatry. 2021;26(10):5481–503. doi: 10.1038/s41380-021-01249-0
- Jessen NA, Munk ASF, Lundgaard I, Nedergaard M. The Glymphatic System: A Beginner’s Guide. Neurochem Res. 2015;40(12):2583–99. doi: 10.1007/s11064-015-1581-6
- Lee H, Xie L, Yu M, Kang H, Feng T, Deane R, et al. The effect of body posture on brain glymphatic transport. J Neurosci. 2015;35(31):11034–44. doi: 10.1523/JNEUROSCI.1625-15.2015
- Morgia C La, Ross-cisneros FN, Koronyo Y, Hannibal J, Gallassi R, Cantalupo G, et al. Melanopsin Retinal Ganglion Cell Loss in Alzheimer Disease. Ann Neurol. 2015;79(1):90–109. doi: 10.1002/ana.24548
- Stern Y. Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol [Internet]. 2012;11(11):1006–12. doi: 10.1016/S1474-4422(12)70191-6
- Fu P, Yung KKL. Air Pollution and Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J Alzheimer’s Dis. 2020;77(2):701–14. doi: 10.3233/JAD-200483
- Livingston G, Huntley J, Sommerlad A, Ames D, Ballard C, Banerjee S, et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet. 2020;396(10248):413–46. doi: 10.1016/S0140-6736(20)30367-6
- Angelopoulou E, Paudel YN, Papageorgiou SG, Piperi C. APOE Genotype and Alzheimer’s Disease: The Influence of Lifestyle and Environmental Factors. ACS Chem Neurosci. 2021;12(15):2749–64. doi: 10.1021/acschemneuro.1c00295
- Berkowitz CL, Mosconi L, Rahman A, Scheyer O, Hristov H, Isaacson RS. Clinical Application of APOE in Alzheimer’s Prevention: A Precision Medicine Approach. J Prev Alzheimer’s Dis. 2018;5(4):245–52. doi: 10.14283/jpad.2018.35
- Gharbi-Meliani A, Dugravot A, Sabia S, Regy M, Fayosse A, Schnitzler A, et al. The association of APOE ε4 with cognitive function over the adult life course and incidence of dementia: 20 years follow-up of the Whitehall II study. Alzheimer’s Res Ther. 2021;13(1):1–11. doi: 10.1186/s13195-020-00740-0
- Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chételat G, Teunissen CE, et al. Alzheimer’s disease. Lancet. 2021;397(10284):1577–90. doi: 10.1016/S0140-6736(20)32205-4
- Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson P V., Bjornsson S, et al. A mutation in APP protects against Alzheimer‘s disease and age-related cognitive decline. Nature. 2012;488(7409):96. doi: 10.1038/nature11283
- Arboleda-Velasquez JF, Lopera F, O’Hare M, Delgado-Tirado S, Marino C, Chmielewska N, et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019 Nov;25(11):1680–3. doi: 10.1038/s41591-019-0611-3
- Lopera F, Marino C, Chandrahas AS, O’Hare M, Villalba-Moreno ND, Aguillon D, et al. Resilience to autosomal dominant Alzheimer’s disease in a Reelin-COLBOS heterozygous man. Nat Med [Internet]. 2023;29(May). doi: 10.1038/s41591-023-02318-3
- Reardon S. How one man’s rare Alzheimer’s mutation delayed the onset of disease. Nature [Internet]. 2023 May 25;617(7962):660–1. doi: 10.1038/d41586-023-01610-z
- Custodio N, Montesinos R, Lira D, Herrera-Pérez E, Bardales Y, Valeriano-Lorenzo L. Demência mista: Revisão das evidências. Dement e Neuropsychol. 2017;11(4):364–70. doi: 10.1590/1980-57642016dn11-040005
- Klijs B, Mitratza M, Harteloh PPM, Moll Van Charante EP, Richard E, Nielen MMJ, et al. Estimating the lifetime risk of dementia using nationwide individually linked cause-of-death and health register data. Int J Epidemiol. 2021;50(3):809–16. doi: 10.1093/ije/dyaa219
- Goldman JS, Hahn SE, Catania JW, Larusse-Eckert S, Butson MB, Rumbaugh M, et al. Genetic counseling and testing for Alzheimer disease: Joint practice guidelines of the American College of Medical Genetics and the National Society of Genetic Counselors. Genet Med. 2011;13(6):597–605. doi: 10.1097/GIM.0b013e31821d69b8
- Mucke HA. The case of galantamine: Repurposing and late blooming of a cholinergic drug. Futur Sci OA. 2015;1(4). doi: 10.4155/fso.15.73
- Pozzi FE, Conti E, Appollonio I, Ferrarese C, Tremolizzo L. Predictors of response to acetylcholinesterase inhibitors in dementia: A systematic review. Front Neurosci. 2022;16. doi: 10.3389/fnins.2022.998224
- Banack SA, Cox PA. Biomagnification of cycad neurotoxins in flying foxes: Implications for ALS-PDC in Guam. Neurology. 2003;61(3):387–9. doi: 10.1212/01.WNL.0000078320.18564.9F
- Sacks OW. L’isola dei senza colore.
- Ross SM, Spencer PS. Specific antagonism of behavioral action of “uncommon” amino acids linked to motor‐system diseases. Synapse. 1987;1(3):248–53. doi: 10.1002/syn.890010305
- Revett TJ, Baker GB, Jhamandas J, Kar S. Glutamate system, amyloid β peptides and tau protein: Functional interrelationships and relevance to Alzheimer disease pathology. J Psychiatry Neurosci. 2013;38(1):6–23. doi: 10.1503/jpn.110190
- Nagata T, Shinagawa S, Inamura K, Shigeta M. Pathogenesis and Personalized Interventions for Pharmacological Treatment-Resistant Neuropsychiatric Symptoms in Alzheimer’s Disease. J Pers Med. 2022;12(9). doi: 10.3390/jpm12091365
- Cummings JL. Defining and labeling disease-modifying treatments for Alzheimer’s disease. Alzheimers Dement. 2009 Sep;5(5):406–18. doi: 10.1016/j.jalz.2008.12.003
- Das B, Yan R. A Close Look at BACE1 Inhibitors for Alzheimer’s Disease Treatment. CNS Drugs [Internet]. 2019;33(3):251–63. doi: 10.1007/s40263-019-00613-7
- Mahase E. Aducanumab : European agency rejects Alzheimer’s drug over efficacy and safety concerns. BMJ. 2021;(375):n3127. doi: 10.1136/bmj.n3127
- Mahase E. Three FDA advisory panel members resign over approval of Alzheimer’s drug. BMJ [Internet]. 2021 Jun 11;372:n1503. doi: 10.1136/bmj.n1503
- Greenberg SM, Bacskai BJ, Hernandez-Guillamon M, Pruzin J, Sperling R, van Veluw SJ. Cerebral amyloid angiopathy and Alzheimer disease — one peptide, two pathways. Nat Rev Neurol [Internet]. 2020 Jan 11;16(1):30–42. doi: 10.1038/s41582-019-0281-2
- Withington CG, Turner RS. Amyloid-Related Imaging Abnormalities With Anti-amyloid Antibodies for the Treatment of Dementia Due to Alzheimer’s Disease. Front Neurol. 2022;13(March). doi: 10.3389/fneur.2022.862369
- Salloway S, Chalkias S, Barkhof F, Burkett P, Barakos J, Purcell D, et al. Amyloid-Related Imaging Abnormalities in 2 Phase 3 Studies Evaluating Aducanumab in Patients with Early Alzheimer Disease. JAMA Neurol. 2022;79(1):13–21. doi: 10.1001/jamaneurol.2021.4161
- Cummings J, Rabinovici GD, Atri A, Aisen P, Apostolova LG, Hendrix S, et al. Aducanumab: Appropriate Use Recommendations Update. J Prev Alzheimer’s Dis. 2022;9(2):221–30. doi: 10.14283/jpad.2022.34
- Padovani A, Caratozzolo S, Rozzini L, Pilotto A, Benussi A, Tedeschi G. “Real-world” eligibility for aducanumab depends on clinical setting and patients’ journey. J Am Geriatr Soc. 2022;70(2):626–8. doi: 10.1111/jgs.17530
- Sinha P, Barocas JA. Cost-effectiveness of aducanumab to prevent Alzheimer’s disease progression at current list price. Alzheimer’s Dement Transl Res Clin Interv. 2022;8(1):8–11. doi: 10.1002/trc2.12256
- Liu J, Hlavka J, Coulter D, Baxi S, Mattke S, Gidengil C. Assessing the Preparedness of the Canadian Health Care System Infrastructure for an Alzheimer’s Treatment. Assess Prep Can Heal Care Syst Infrastruct an Alzheimer’s Treat. 2019; doi: 10.7249/rr2744
- Hlavka JP, Mattke S, Liu JL. Assessing the Preparedness of the Health Care System Infrastructure in Six European Countries for an Alzheimer’s Treatment. Rand Heal Q [Internet]. 2019 May;8(3):2. doi: 10.1007/978-1-4939-6556-4_7
- van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, et al. Lecanemab in Early Alzheimer’s Disease. N Engl J Med [Internet]. 2023 Jan 5;388(1):9–21. doi: 10.1056/NEJMoa2212948
- Klunk WE, Koeppe RA, Price JC, Benzinger TL, Devous MD, Jagust WJ, et al. The Centiloid project: Standardizing quantitative amyloid plaque estimation by PET. Alzheimer’s Dement [Internet]. 2015;11(1):1-15.e4. doi: 10.1016/j.jalz.2014.07.003
- Chalkias S, Mummery CJ, Salloway S, Barkhof F, Burkett P, Purcell D, et al. Evaluation of aducanumab safety in early Alzheimer ’ s disease. 2021;1–16.
- Barkhof F, Klein G, Tonietto M, Bittner T, Ahlers S, Abbas R. GRADUATE I and II : Imaging Findings From Two Phase III Trials of Subcutaneous Gantenerumab in Early Alzheimer ’ s Disease. 2023;1–21.
- Tousi B. Antiamyloid Antibody Therapy in Alzheimer Disease. Pract Neurol. 2022;(August):63–6.
- Söderberg L, Johannesson M, Nygren P, Laudon H, Eriksson F, Osswald G, et al. Lecanemab, Aducanumab, and Gantenerumab — Binding Profiles to Different Forms of Amyloid-Beta Might Explain Efficacy and Side Effects in Clinical Trials for Alzheimer’s Disease. Neurotherapeutics [Internet]. 2022;20(1):195–206. doi: 10.1007/s13311-022-01308-6
- Wischik CM, Bentham P, Gauthier S, Miller S, Kook K, Schelter BO. Oral Tau Aggregation Inhibitor for Alzheimer’s Disease: Design, Progress and Basis for Selection of the 16 mg/day Dose in a Phase 3, Randomized, Placebo-Controlled Trial of Hydromethylthionine Mesylate. J Prev Alzheimer’s Dis [Internet]. 2022; doi: 10.14283/jpad.2022.63
- Press Release: LUCIDITY phase 3 topline data presented at CTAD for HMTM – the only oral anti-Tau therapy in late-stage development. 2022;1–2.
- Reardon S. Alzheimer’s drug donanemab: what promising trial means for treatments. Nature [Internet]. 2023 May 11;617(7960):232–3. doi: 10.1038/d41586-023-01537-5
- Tahami Monfared AA, Tafazzoli A, Chavan A, Ye W, Zhang Q. The Potential Economic Value of Lecanemab in Patients with Early Alzheimer’s Disease Using Simulation Modeling. Neurol Ther [Internet]. 2022;11(3):1285–307. doi: 10.1007/s40120-022-00373-5
- Ross EL, Weinberg MS, Arnold SE. Cost-effectiveness of Aducanumab and Donanemab for Early Alzheimer Disease in the US. JAMA Neurol. 2022;79(5):478–87. doi: 10.1001/jamaneurol.2022.0315
- Gauthier S, Ismail Z, Goodarzi Z, Ng KP, Rosa-Neto P. Clinicians’ Perspectives on How Disease Modifying Drugs for Alzheimer’s Disease Impact Specialty Care. J Prev Alzheimer’s Dis. 2023;3(10):339–41. doi: 10.14283/jpad.2023.72
- Sampaio C. «Compounded Interest» in Alzheimer’s Disease: Do New Amyloid-Targeting Treatments Justify Their Use. J Prev Alzheimer’s Dis. 2023;3(10):333–5. doi: 10.14283/jpad.2023.70
- Schneider LS. When It Comes to Lecanemab (and Donanemab), How Might We Think about ‘Reasonable and Necessary’? J Prev Alzheimer’s Dis. 2023;3(10):342–3. doi: 10.14283/jpad.2023.73
- Iwatsubo T. 2022 Clarity AD CTAD Presentations. Ctad 2022. 2022;
- Rafii MS, Sperling RA, Donohue MC, Zhou J, Roberts C, Irizarry MC, et al. The AHEAD 3-45 Study: Design of a prevention trial for Alzheimer’s disease. Alzheimer’s Dement. 2022;(June 2022):1227–33. doi: 10.1002/alz.12748
- Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement [Internet]. 2018;14(4):535–62. doi: 10.1016/j.jalz.2018.02.018
- Solomon A, Turunen H, Ngandu T, Peltonen M, Levälahti E, Helisalmi S, et al. Effect of the apolipoprotein e genotype on cognitive change during a multidomain lifestyle intervention a subgroup analysis of a randomized clinical trial. JAMA Neurol [Internet]. 2018;75(4):462–70. doi: 10.1001/jamaneurol.2017.4365
- Kivipelto M, Ngandu T, Laatikainen T, Winblad B, Soininen H, Tuomilehto J. Risk score for the prediction of dementia risk in 20 years among middle aged people: a longitudinal, population-based study. Lancet Neurol. 2006;5(9):735–41. doi: 10.1016/S1474-4422(06)70537-3
- Ngandu T, Lehtisalo J, Solomon A, Levälahti E, Ahtiluoto S, Antikainen R, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): A randomised controlled trial. Lancet [Internet]. 2015; doi: 10.1016/S0140-6736(15)60461-5 LK – https://unimib.on.worldcat.org/atoztitles/link?sid=EMBASE&sid=EMBASE&issn=1474547X&id=doi:10.1016%2FS0140-6736%2815%2960461-5&atitle=A+2+year+multidomain+intervention+of+diet%2C+exercise%2C+cognitive+training%2C+and+vascular+risk+monitoring+versus+control+to+prevent+cognitive+decline+in+at-risk+elderly+people+%28FINGER%29%3A+A+randomised+controlled+trial&stitle=Lancet&title=The+Lancet&volume=&issue=&spage=&epage=&aulast=Ngandu&aufirst=Tiia&auinit=T.&aufull=Nga
- Rosenberg A, Mangialasche F, Ngandu T, Solomon A, Kivipelto M. Multidomain Interventions to Prevent Cognitive Impairment, Alzheimer’s Disease, and Dementia: From FINGER to World-Wide FINGERS. J Prev Alzheimer’s Dis. 2020;7(1):29–36. doi: 10.14283/jpad.2019.41
- Kivipelto M, Mangialasche F, Snyder HM, Allegri R, Andrieu S, Arai H, et al. World-Wide FINGERS Network: A global approach to risk reduction and prevention of dementia. Alzheimer’s Dement. 2020;16(7):1078–94. doi: 10.1002/alz.12123
- Morris MC, Tangney CC, Wang Y, Sacks FM, Bennett DA, Aggarwal NT. MIND diet associated with reduced incidence of Alzheimer’s disease. Alzheimer’s Dement [Internet]. 2015;11(9):1007–14. doi: 10.1016/j.jalz.2014.11.009
- Morris MC, Tangney CC, Wang Y, Sacks FM, Barnes LL, Bennett DA, et al. MIND diet slows cognitive decline with aging. Alzheimer’s Dement [Internet]. 2015;11(9):1015–22. doi: 10.1016/j.jalz.2015.04.011
- Jessen F, Amariglio RE, Van Boxtel M, Breteler M, Ceccaldi M, Chételat G, et al. A conceptual framework for research on subjective cognitive decline in preclinical Alzheimer’s disease. Alzheimer’s Dement. 2014;10(6):844–52. doi: 10.1016/j.jalz.2014.01.001
- Slot RER, Sikkes SAM, Berkhof J, Brodaty H, Buckley R, Cavedo E, et al. Subjective cognitive decline and rates of incident Alzheimer’s disease and non–Alzheimer’s disease dementia. Alzheimer’s Dement. 2019;15(3):465–76. doi: 10.1016/j.jalz.2018.10.003
- Slot RER, Verfaillie SCJ, Overbeek JM, Timmers T, Wesselman LMP, Teunissen CE, et al. Subjective Cognitive Impairment Cohort (SCIENCe): Study design and first results. Alzheimer’s Res Ther. 2018;10(1):1–13. doi: 10.1186/s13195-018-0390-y
- Jessen F, Amariglio RE, Buckley RF, van der Flier WM, Han Y, Molinuevo JL, et al. The characterisation of subjective cognitive decline. LANCET Neurol. 2020;19(3):271–8. doi: 10.1016/S1474-4422(19)30368-0
- Röhr S, Pabst A, Riedel-Heller SG, Jessen F, Turana Y, Handajani YS, et al. Estimating prevalence of subjective cognitive decline in and across international cohort studies of aging: a COSMIC study. Alzheimer’s Res Ther [Internet]. 2020;12(1). doi: 10.1186/s13195-020-00734-y
- CDC. Subjective cognitive decline — a public health issue. US Dep Heal Hum Serv [Internet]. 2018;10(6):844–52.
- Mukadam N, Sommerlad A, Huntley J, Livingston G. Population attributable fractions for risk factors for dementia in low-income and middle-income countries: an analysis using cross-sectional survey data. Lancet Glob Heal [Internet]. 2019;7(5):e596–603. doi: 10.1016/S2214-109X(19)30074-9
- van Maurik IS, Visser LNC, Pel-Littel RE, van Buchem MM, Zwan MD, Kunneman M, et al. Development and usability of ADappt: Web-based tool to support clinicians, patients, and caregivers in the diagnosis of mild cognitive impairment and alzheimer disease. JMIR Form Res [Internet]. 2019;3(3). doi: 10.2196/13417
- Altomare D, Molinuevo JL, Ritchie C, Ribaldi F, Carrera E, Dubois B, et al. Brain Health Services: organization, structure, and challenges for implementation. A user manual for Brain Health Services—part 1 of 6. Alzheimer’s Res Ther. 2021;13(1):1–11. doi: 10.1186/s13195-021-00827-2
- Ranson JM, Rittman T, Hayat S, Brayne C, Jessen F, Blennow K, et al. Modifiable risk factors for dementia and dementia risk profiling. A user manual for Brain Health Services-part 2 of 6. Alzheimers Res Ther. 2021 Oct;13(1):169. doi: 10.1186/s13195-021-00895-4
- Visser LNC, Minguillon C, Sánchez-Benavides G, Abramowicz M, Altomare D, Fauria K, et al. Dementia risk communication. A user manual for Brain Health Services—part 3 of 6. Alzheimer’s Res Ther. 2021;13(1):1–12. doi: 10.1186/s13195-021-00840-5
- Solomon A, Stephen R, Altomare D, Carrera E, Frisoni GB, Kulmala J, et al. Multidomain interventions: state-of-the-art and future directions for protocols to implement precision dementia risk reduction. A user manual for Brain Health Services—part 4 of 6. Alzheimer’s Res Ther. 2021;13(1):1–15. doi: 10.1186/s13195-021-00875-8
- Brioschi Guevara A, Bieler M, Altomare D, Berthier M, Csajka C, Dautricourt S, et al. Protocols for cognitive enhancement. A user manual for Brain Health Services—part 5 of 6. Alzheimer’s Res Ther. 2021;13(1):1–13. doi: 10.1186/s13195-021-00844-1
- Milne R, Altomare D, Ribaldi F, Molinuevo JL, Frisoni GB, Brayne C. Societal and equity challenges for Brain Health Services. A user manual for Brain Health Services—part 6 of 6. Alzheimer’s Res Ther. 2021;13(1):1–8. doi: 10.1186/s13195-021-00885-6
- Frisoni GB, Altomare D, Ribaldi F, Villain N, Brayne C, Mukadam N, et al. Dementia prevention in memory clinics: recommendations from the European task force for brain health services. Lancet Reg Heal – Eur. 2023;26:1–15. doi: 10.1016/j.lanepe.2022.100576
© COPYRIGHT Illustrazione di Raffaella Cocchi per BRAINFACTOR Tutti i diritti riservati.
Leave a comment